Quantitative Mass Spectrometry Based Proteomics for Characterizing Modified Proteins and Proteomes
The sequences of the human genome and genomes of many other organisms are now readily available and have revolutionized modern biological research. Nevertheless, the next challenge presently on the horizon (after the post-genome era) is the comprehensive characterization of cellular proteins (i.e. the “proteome”), the ‘active/expressed’ part of the genome. DNA sequence or mRNA levels alone cannot predict the dynamic aspects of cellular function. Proteins, their post-translational modifications (PTMs) and the multi-protein complexes they form are the  driving forces of cellular machinery that control a diverse number of physiological events. These observations have led to the emergence of a new sub-field of contemporary biology called Proteomics: the characterization of the protein complement expressed by a genome of a particular organism, tissue or cell. At the heart of proteomic experiments is the use of nanoflow liquid chromatography-tandem mass spectrometry for the analysis of complex protein mixtures, which is arguably the most rapid, sensitive and accurate technique available for sequence characterization of proteins.
driving forces of cellular machinery that control a diverse number of physiological events. These observations have led to the emergence of a new sub-field of contemporary biology called Proteomics: the characterization of the protein complement expressed by a genome of a particular organism, tissue or cell. At the heart of proteomic experiments is the use of nanoflow liquid chromatography-tandem mass spectrometry for the analysis of complex protein mixtures, which is arguably the most rapid, sensitive and accurate technique available for sequence characterization of proteins.
The Garcia Lab is focused on developing novel mass spectrometry based proteomic methodologies for quantitatively characterizing changes in protein and proteome expression and post-translational modification state during significant biological events, or in response to external perturbation. Our goal is to utilize large-scale proteomic data to improve our understanding of biological processes at the molecular level. Work in the lab is centered on continued innovation in proteomic technology applied to several areas of cellular biology.
Towards deciphering the epigenetic Histone Code
Epigenetics refers to stable heritable changes in gene expression that are not due to changes in DNA sequence, such as DNA methylation, RNA interference and histone PTMs. These epigenetic changes are responsible for generating different cell types originating from the exact same genome. Emerging as one key regulator of cellular memory are histones. Histones are small basic proteins that function to package genomic DNA into repeating nucleosomal units (containing ~146 bp of DNA wrapped around two copies each of histones H3, H4, H2A and H2B) forming the chromatin fiber and hence our chromosomes. In general, the packaging of DNA into chromatin is recognized to be a major mechanism by which the access of genomic DNA is restricted. This physical barrier to the underlying DNA is precisely regulated, at least in part, by the PTMs on histones.
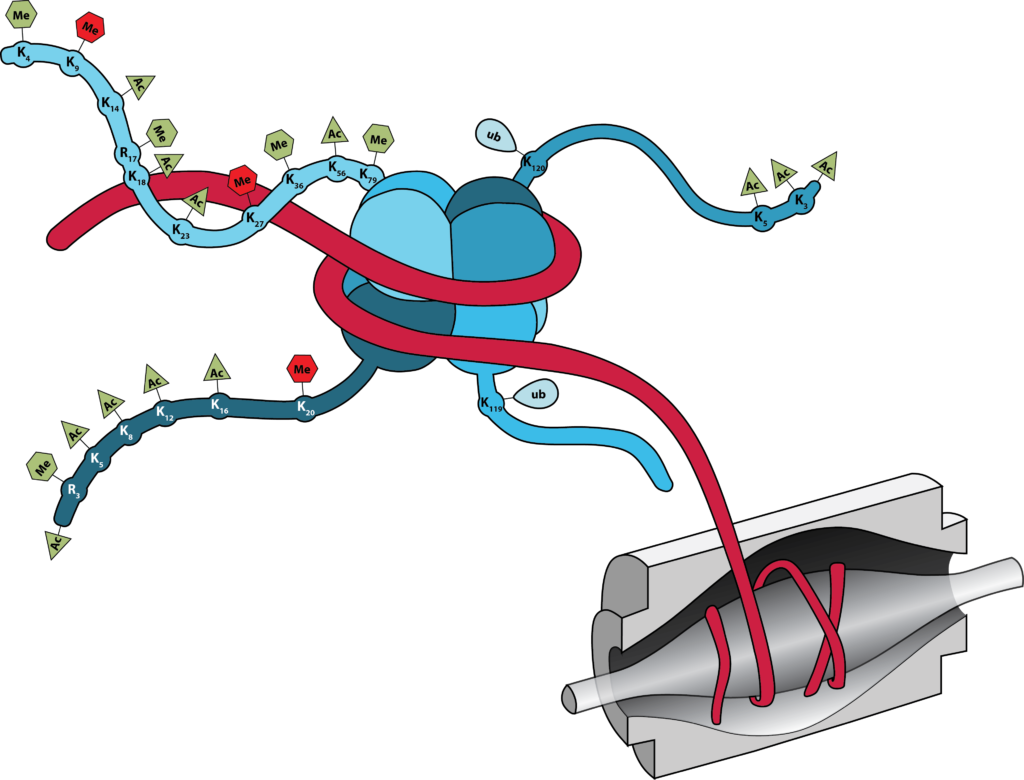 A wide number of studies show that several single covalent histone modifications such as methylation, acetylation, phosphorylation and ubiquitination located in the N-terminal tails correlate with both the regulation of chromatin structure during active gene expression, or heterochromatin formation during gene silencing (i.e. the “Histone Code”). Nevertheless, it is currently unknown what effects, if any, multiple combinations of histone modifications might exert, and translating the combinatorial modification patterns of histones into biological significance remains a significant challenge. These histone PTMs occur on multiple but specific sites, suggesting that histones can act as signaling platforms for proteins that bind or “read” these marks. In support, several proteins that contain special domains that bind various PTM sites on histones have been discovered. The Garcia Lab has developed arguably the best precision mass spectrometry based platform for detecting and quantifying the combinatorial histone PTM code.
A wide number of studies show that several single covalent histone modifications such as methylation, acetylation, phosphorylation and ubiquitination located in the N-terminal tails correlate with both the regulation of chromatin structure during active gene expression, or heterochromatin formation during gene silencing (i.e. the “Histone Code”). Nevertheless, it is currently unknown what effects, if any, multiple combinations of histone modifications might exert, and translating the combinatorial modification patterns of histones into biological significance remains a significant challenge. These histone PTMs occur on multiple but specific sites, suggesting that histones can act as signaling platforms for proteins that bind or “read” these marks. In support, several proteins that contain special domains that bind various PTM sites on histones have been discovered. The Garcia Lab has developed arguably the best precision mass spectrometry based platform for detecting and quantifying the combinatorial histone PTM code.
The Garcia Lab has developed several proteomics approaches that are considered state of the art for histone PTM analyses and are used world-wide by many research groups. For example, recently we have been leading the application of data independent acquisition (DIA) mass spectrometry for enhanced quantification of histone PTMs, especially isomeric PTM modified species. Therefore, we believe that the utilization of advanced proteomic technology in the chromatin biology field will enhance investigations of histone modifications to a much higher scale. In combination with cell and biochemical experimentation, bioinformatics analysis and other “omics” technologies; our large-scale proteomic data will help provide a systems biology outlook on epigenetic processes that will lay the foundation for development of drug treatments for human diseases that are believed to involve epigenetic mechanisms.
Dynamics of proteome-wide PTM mediated biological pathways
Another goal of the Garcia Lab is to develop and apply novel proteomics based methodology to understand how signaling pathways affect cellular functions and ultimately cell phenotypes. We use quantitative mass spectrometry to measure dynamic changes in protein abundances, protein PTM states, and to characterize protein:protein interactions. For example, we are specifically developing large-scale approaches to isolate and characterize a variety of different types of PTM modified proteomes (e.g. protein phosphorylation, methylation, acetylation, glycosylation, ADP-ribosylation, etc.).
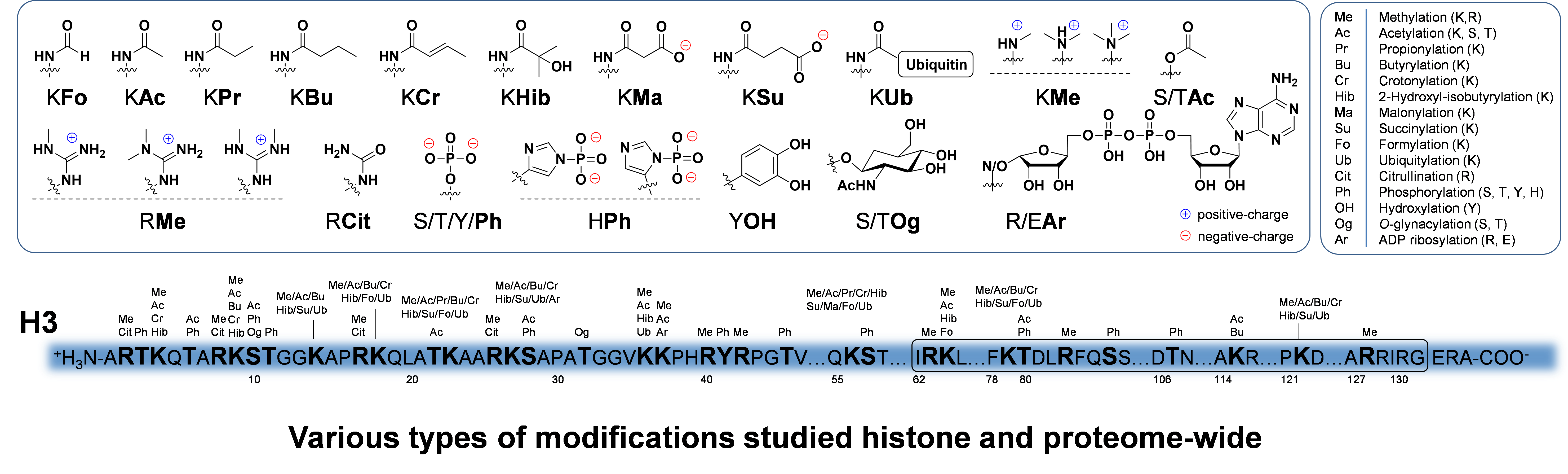 These types of approaches allow for example, the detection of thousands of modified proteins from cells or tissues. When combined with quantitative proteomics techniques such as stable isotope labeling of amino acids in cell culture (SILAC), these cutting-edge tools allow us to examine with unprecedented detail, the molecular level events involved in various biological processes or disease state. We are also very interested in the dynamics of protein modification, and have developed in vivo cellular metabolic labeling strategies to specifically label newly modified proteins. For example, if we label mammalian cells with stable isotopically heavy 13C-glucose, we will then detect the 13C tracer on carbons from acetyl-CoA and UDP-GlcNAc which will ultimately end up on acetylated and GlcNAcylated proteins. These experiments have allowed us to define for the first time the dynamics/half-lives of particular classes of modified proteins on a proteome-scale. In addition, this methodology allows us to determine protein modification kinetics in response to external stimuli such as drug/inhibitor treatments, or to understand the link between metabolism and the epigenome/proteome. Lastly, we are extremely interested in how these different protein PTMs affect one another, and we are developing the MS platforms to determine which modifications are found simultaneously on the same proteins, and how this biological code is then transformed to direct a myriad of cellular functions.
These types of approaches allow for example, the detection of thousands of modified proteins from cells or tissues. When combined with quantitative proteomics techniques such as stable isotope labeling of amino acids in cell culture (SILAC), these cutting-edge tools allow us to examine with unprecedented detail, the molecular level events involved in various biological processes or disease state. We are also very interested in the dynamics of protein modification, and have developed in vivo cellular metabolic labeling strategies to specifically label newly modified proteins. For example, if we label mammalian cells with stable isotopically heavy 13C-glucose, we will then detect the 13C tracer on carbons from acetyl-CoA and UDP-GlcNAc which will ultimately end up on acetylated and GlcNAcylated proteins. These experiments have allowed us to define for the first time the dynamics/half-lives of particular classes of modified proteins on a proteome-scale. In addition, this methodology allows us to determine protein modification kinetics in response to external stimuli such as drug/inhibitor treatments, or to understand the link between metabolism and the epigenome/proteome. Lastly, we are extremely interested in how these different protein PTMs affect one another, and we are developing the MS platforms to determine which modifications are found simultaneously on the same proteins, and how this biological code is then transformed to direct a myriad of cellular functions.
Comprehensive characterization of crosstalk between cellular signaling pathways and epigenetic networks
Protein phosphorylation mediated signaling events play significant roles in most biological processes including cellular differentiation, proliferation and apoptosis. The Garcia Lab is also highly interested in understanding a long-standing unresolved question in the chromatin biology field: How do signal transduction pathways directly communicate from the outer cell surface into the nucleus to ultimately remodel chromatin protein networks and influence gene expression? To tackle this question, we combine the use of several approaches including proteomics, genomics, chemical biology, etc. To characterize protein phosphorylation, we employ phosphopeptide enrichment strategies in combination with nanoLC-MS/MS to quantify >5000 phosphorylation sites from minute amounts of total cell lysate. By employing biochemical fractionation (i.e. cytoplasmic, nuclear and chromatin) with the above approach, we can begin to understand the signaling pathways utilized and how these signals are relayed into chromatin to phosphorylate and activate chromatin remodeling proteins, epigenetic machinery or transcription factors .
We have also been developing chemical biology and genomic methods for investigating the landscape of sub chromatin-wide portions of genome. In order to identify direct substrates of activated pathways and kinases, we have employed modified ATP analogs to specifically mark and allow for the enrichment of kinase targets on chromatin. This allows us to determine the potential mechanisms for transcriptional activation, and also to use next generation genome sequencing approaches to identify kinase regulated target genes. There are also several efforts in the lab to combine genomic technologies with proteomic readouts to define local chromatin environments. This can be accomplished using various chromatin immunoprecipitation (ChIP) or reverse-ChIP sample preparations with sensitive quantitative proteomic measurements. Major interests include comprehensive characterization of genomic locations harboring specific histone modifications or variants, chromatin binding proteins (i.e. histone PTM readers), or other chromatin associated proteins (i.e. transcription factors). The overall goals of these studies are to take a defined part of the genome and accurately quantify the dynamic their dynamic content (histone PTMs and non-histone proteins), thereby taking a snapshot of the chromatin landscape.
Understanding cellular and epigenetic mechanisms involved in normal and disease physiology
Histone PTM mediated epigenetic mechanisms have shown to have incredible roles in human biology and disease. For example, growing evidence has linked alterations to epigenetic mechanisms with various human diseases such as autoimmune, neurological disorders and cancer. These observations have made a strong case for the initiation of an international Human Epigenome Project on the scale of the Human Genome Project. Understanding epigenetic changes in development and disease would make immediate impacts in many biomedical areas ranging from regenerative medicine, aging, reproduction, cancer, and neuroscience; and could serve as a novel means of disease targeted therapeutic intervention for some specific human diseases. A selected few of the Garcia Lab’s main biological/biomedical interests are described below:
A first area of interest is in understanding the role of the epigenome in stem cell pluripotency and differentiation. Both undifferentiated and differentiated embryonic stem cells (ESCs) are widely used to investigate epigenetic regulation. The former because they express a large range of epigenetic modifying enzymes, and the latter because they serve as an excellent model for dynamic chromatin remodeling and epigenetic regulation of gene expression. As such, epigenetic mechanisms are felt to play large roles in ESC physiology and control of gene expression patterns key to cellular differentiation. We currently have various stem cell model systems where we are applying quantitative proteomics and genomics experiments to understand how combinatorial histone PTMs, signaling pathways, chromatin modifying enzymes and histone PTM readers direct differentiation of pluripotent stem cells to specific lineages.
 Another research interest is to examine how viruses hijack epigenetic mechanisms to allow for their survival and replication. Upon infection of cells, some DNA viruses interact with many nuclear proteins (such as histones) becoming “chromatinized” and ultimately harnessing host epigenetic machinery to modulate both viral and cellular gene expression. Here we aim to answer the following questions: How do viruses utilize histone PTMs for controlling both host and viral gene expression patterns and replication? Are there any non-histone chromatin proteins that are needed for viral growth? As certain protein lysine methyltransferases are upregulated during viral infection, are there other non-histone protein modifications such as methylation that are vital to this process? We hope to parse out the epigenetic changes on both host cellular chromatin, as well as on chromatinized viral DNA using a multitude of strategic quantitative methods.
Another research interest is to examine how viruses hijack epigenetic mechanisms to allow for their survival and replication. Upon infection of cells, some DNA viruses interact with many nuclear proteins (such as histones) becoming “chromatinized” and ultimately harnessing host epigenetic machinery to modulate both viral and cellular gene expression. Here we aim to answer the following questions: How do viruses utilize histone PTMs for controlling both host and viral gene expression patterns and replication? Are there any non-histone chromatin proteins that are needed for viral growth? As certain protein lysine methyltransferases are upregulated during viral infection, are there other non-histone protein modifications such as methylation that are vital to this process? We hope to parse out the epigenetic changes on both host cellular chromatin, as well as on chromatinized viral DNA using a multitude of strategic quantitative methods.
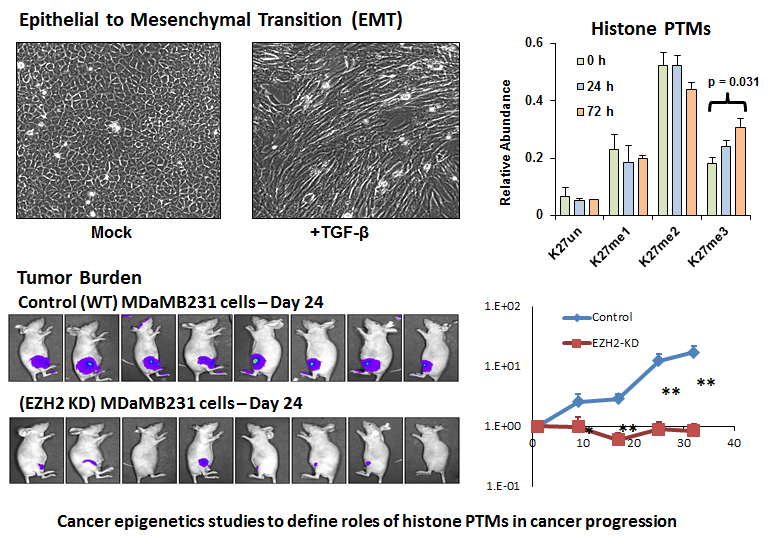
Lastly, we are also highly motivated to examine the roles of epigenetic mechanisms during cancer progression. Cancer is a disease that consists of both genetic and epigenetic alterations. Recently, increasing evidence demonstrates that the genetic changes found in cancer patients often results in gain or loss of function of chromatin enzymes and factors leading to misregulated epigenome patterns. Current projects in this area in the lab include profiling epigenetic histone modifications during important initiation of metastasis such as the epithelial to mesenchymal transition. Additionally, we are investigating “oncogenic” histone H3 mutations that have been discovered in several cancers including high-grade gliomas. These oncohistone mutations often occur at residues that are close to or hot spots for modification, such as the H3K27 to M mutation (H3K27M). We are taking a holistic approach to comprehensively determine how these oncohistone mutations influence modifications on nearby residues, and also consequently affect gene expression patterns that disrupt proteome and signaling pathway homeostasis.
We thank all of our current and previous funding sources!
